Welche Rolle spielen Gene bei der Entstehung einer SMA?
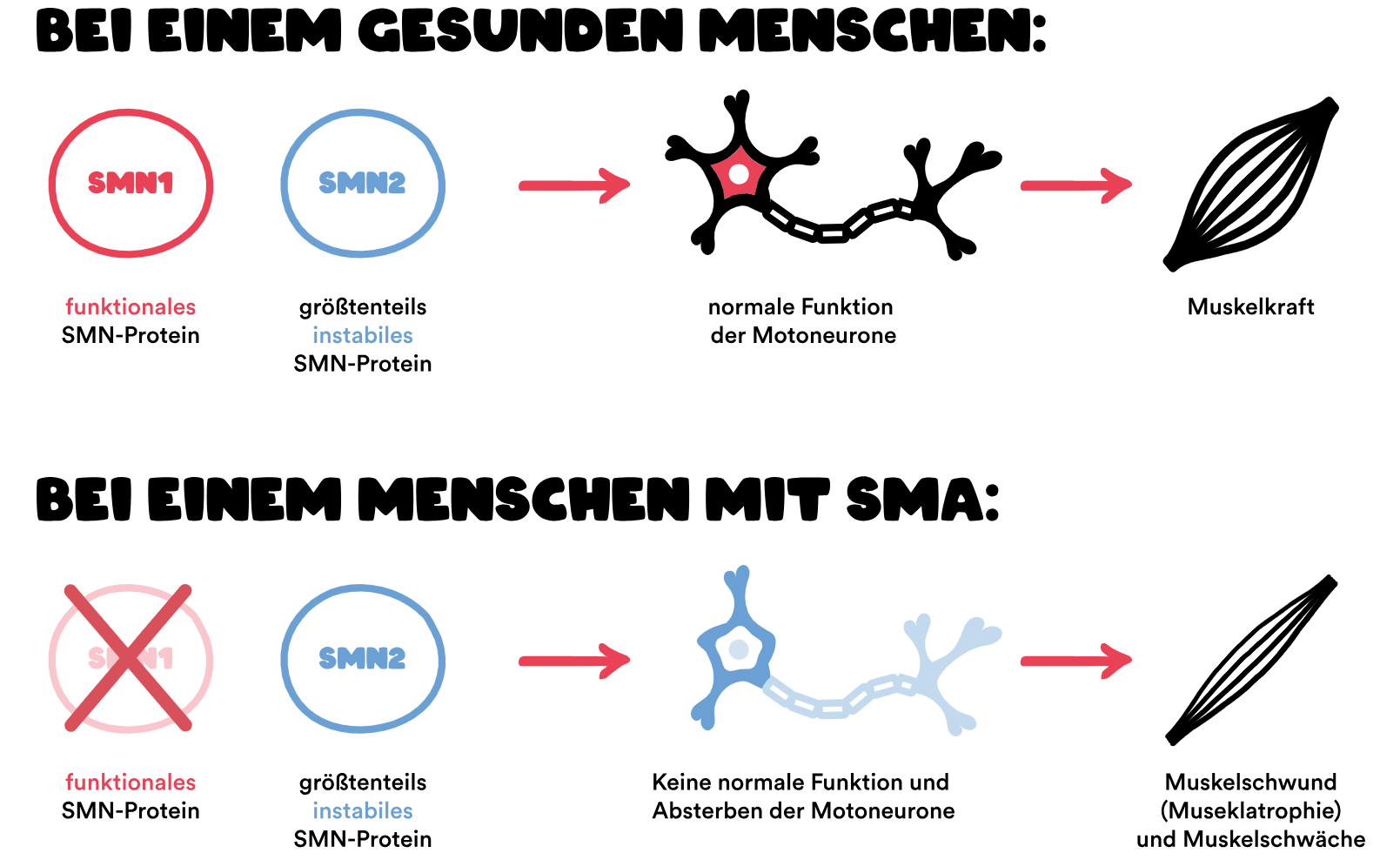
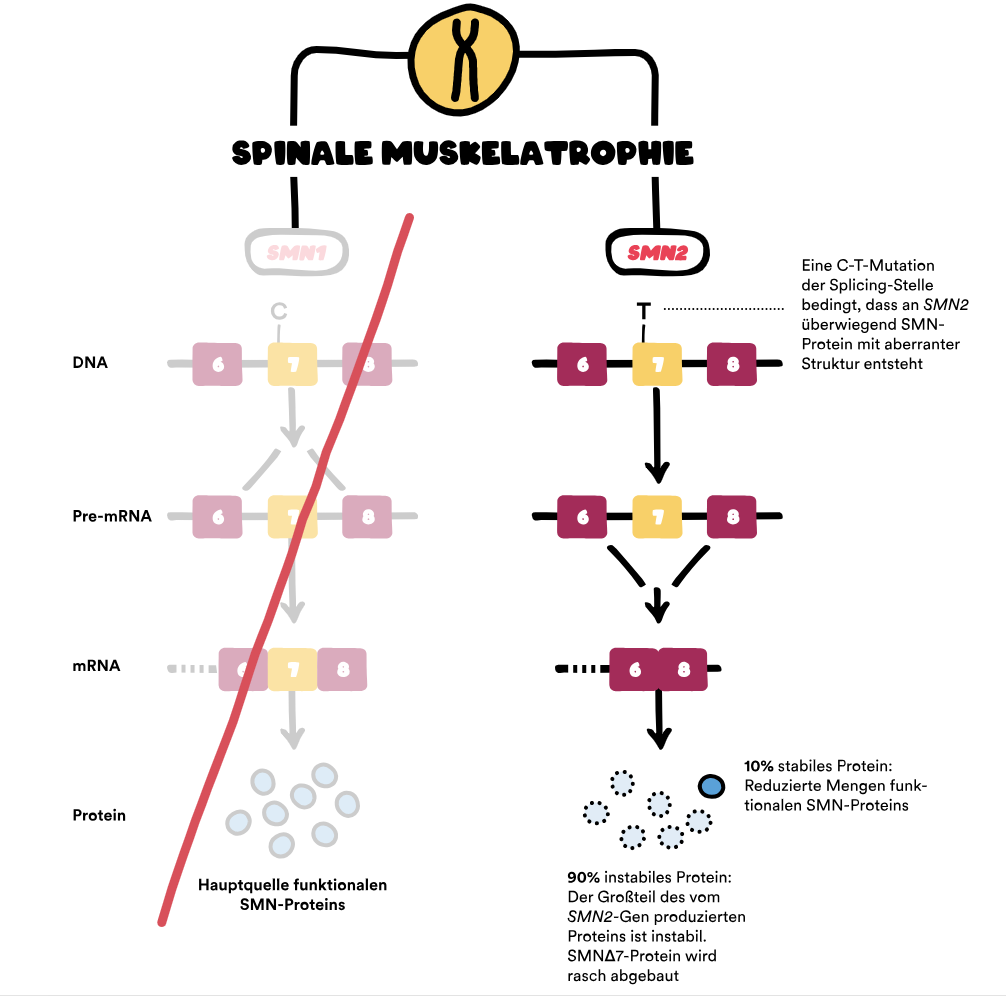
Gene spielen eine zentrale Rolle bei der Spinalen Muskelatrophie (SMA). Die Ursache von SMA ist ein Gendefekt, bei dem das Survival-of-Motor-Neuron-Gen (SMN1-Gen) fehlt (Deletion) oder eine Veränderung (Mutation) aufweist. Dieses Gen liegt auf dem q-Arm des Chromosoms 5 und sorgt für die Bildung des SMN-Proteins. Deswegen spricht man auch von 5q-assoziierter SMA – sie ist die häufigste Form der SMA. 1
Bei 96 Prozent aller Menschen mit 5q-assoziierter SMA liegt ein beidseitiger (homozygoter) Verlust des SMN1-Gens vor. 2 Die anderen 4 Prozent weisen stattdessen eine komplexe Heterozygotie auf: Nur eins der beiden SMN1-Gene fehlt, das andere ist so verändert, dass es kein funktionsfähiges SMN-Protein herstellen kann (Loss-of-function-Mutation). 2